Review Article
Transglutaminase inhibition: possible therapeutic mechanisms to protect cells from death in neurological disorders

Rosaria Romano1, Nicola Gaetano Gatta2, Elenamaria Fioretti2 and Vittorio Gentile2*
1Department of Experimental Medicine, University of Campania “Luigi Vanvitelli”, via Costantinopoli 16, 80138 Naples, Italy
2Department of Biochemistry, Biophysics and General Pathology, Italy
*Address for Correspondence: Dr. Vittorio Gentile, Department of Biochemistry, Biophysics and General Pathology, Italy, Tel: +0039-81-5665870; Fax: +0039-81-5665863; Email: [email protected]
Dates: Submitted: 22 June 2017; Approved: 24 July 2017; Published: 25 July 2017
How to cite this article: Romano R, Gatta NG, Fioretti E, Gentile V. Transglutaminase inhibition: possible therapeutic mechanisms to protect cells from death in neurological disorders. Insights Biol Med. 2017; 1: 026-038. DOI: 10.29328/journal.ibm.1001004
Copyright License: © 2017 Romano R, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Transglutaminases; Post-translational Modifications of Proteins; Neurodegeneration; NF-kB; Neuroinflammation
ABSTRACT
Transglutaminases are a family of Ca2+-dependent enzymes which catalyze post-translational modifications of proteins. The main activity of these enzymes is the cross-linking of glutaminyl residues of a protein/peptide substrate to lysyl residues of a protein/peptide co-substrate. In addition to lysyl residues, other second nucleophilic co-substrates may include monoamines or polyamines (to form mono-or bi-substituted/crosslinked adducts) or -OH groups (to form ester linkages). In absence of co-substrates, the nucleophile may be water, resulting in the net deamidation of the glutaminyl residue. Transglutaminase activity has been suggested to be involved in molecular mechanisms responsible for both physiological and pathological processes. In particular, transglutaminase activity has been shown to be responsible for human autoimmune diseases, and Celiac Disease is just one of them. Interestingly, neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, supranuclear palsy, Huntington’s disease and other polyglutamine diseases, are characterized in part by aberrant cerebral transglutaminase activity and by increased cross-linked proteins in affected brains. Here we describe the possible molecular mechanisms by which these enzymes could be responsible for such diseases and the possible use of transglutaminase inhibitors for patients with diseases characterized by aberrant transglutaminase activity.
BIOCHEMISTRY OF THE TRANSGLUTAMINASES
Transglutaminases (TGs, E.C.2.3.2.13) are Ca2+-dependent enzymes which catalyze post-translational modifications of proteins. Examples of TG-catalyzed reactions include: (I) acyl transfer between the γ-carboxamide group of a protein/polypeptide glutaminyl residue and the ε-amino group of a protein/polypeptide lysyl residue; (II) attachment of a polyamine to the γ-carboxamide of a glutaminyl residue; (III) deamidation of the γ-carboxamide group of a protein/polypeptide glutaminyl residue (Figure 1) [1,2]. The reactions catalyzed by TGs occur by a two-step mechanism (ping-pong type), (Figure 2). The transamidating activity of TGs is activated by the binding of Ca2+, which exposes an active-site cysteine residue. This cysteine residue reacts with the γ-carboxamide group of an incoming glutaminyl residue of a protein/peptide substrate to yield a thioacyl-enzyme intermediate and ammonia, (Figure 2, Step 1). The thioacyl-enzyme intermediate then reacts with a nucleophilic primary amine substrate, resulting in the covalent attachment of the amine-containing donor to the substrate glutaminyl acceptor and regeneration of the cysteinyl residue at the active site, (Figure 2, Step 2). If the primary amine is donated by the ε-amino group of a lysyl residue in a protein/polypeptide, a Nε-(γ-L-glutamyl)-L-lysine (GGEL) isopeptide bond is formed, (Figure 1A, example I). On the other hand, if a polyamine or another primary amine (e.g. histamine, serotonin and others) acts as the amine donor, a γ-glutamylpolyamine (or γ-glutamylamine) residue is formed, (Figure 1, example II). It is also possible for a polyamine to act as an N,N-bis-(γ-L-glutamyl)polyamine bridge between two glutaminyl acceptor residues either on the same protein/polypeptide or between two proteins/polypeptides [3]. If there is no primary amine present, water may act as the attacking nucleophile, resulting in the deamidation of glutaminyl residues to glutamyl residues, (Figure 1, example III). Regarding the physiological roles played by the transglutaminase activity, recently transglutaminase-catalyzed polyamination of tubulin has been shown to stabilize axonal microtubules, suggesting an important role for these reactions also during some physiological processes, such as neurite outgrowth and axon maturation [4]. The reactions catalyzed by TGs occur with little change in free energy and hence should theoretically be reversible. However, under physiological conditions the cross linking reactions catalyzed by TGs are usually irreversible. This irreversibility partly results from the metabolic removal of ammonia from the system and from thermodynamic considerations resulting from altered protein conformation. Some scientific reports suggest that TGs may be able to catalyze the hydrolysis of Nε-(γ-L-glutamyl)-L-lysine cross-links (GGEL) isopeptide bonds in some soluble cross-linked proteins. Furthermore, it is likely that TGs can catalyze the exchange of polyamines onto proteins [2]. In TG2 other catalytic activities, such as the ability to hydrolyze GTP (or ATP) into GDP (or ADP) and inorganic phosphate (Figure 1, example IV), a protein disulfide isomerase activity (Figure 1, example V), and a kinase activity which phosphorylates histones, retinoblastoma (RB) and P53 (Figure 1, example VI), are present, while only some of these activities have been identified also in other TGs [5-8].
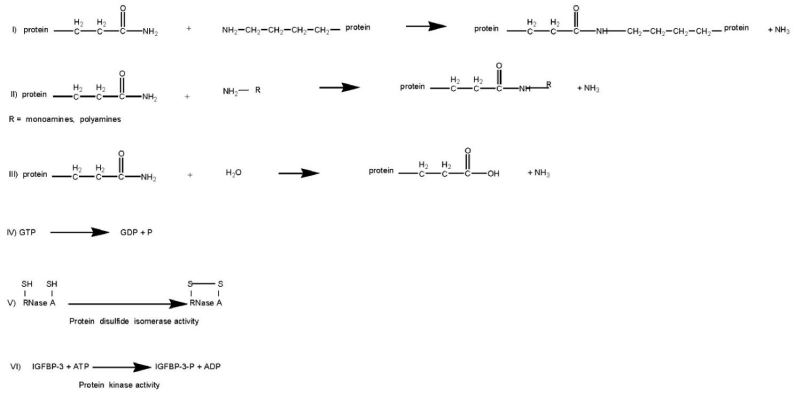
Figure 1: Examples of reactions catalyzed by TG: (I) acyl transfer between the γ-carboxamide group of a protein/polypeptide glutaminyl residue and the ε-amino group of a protein/polypeptide lysyl residue; (II) attachment of a polyamine to the γ-carboxamide of a glutaminyl residue; (III) deamidation of the γ-carboxamide group of a protein/polypeptide glutaminyl residue; (IV) GTPase activity; (V) protein disulfide isomerase activity; (VI) protein kinase activity.
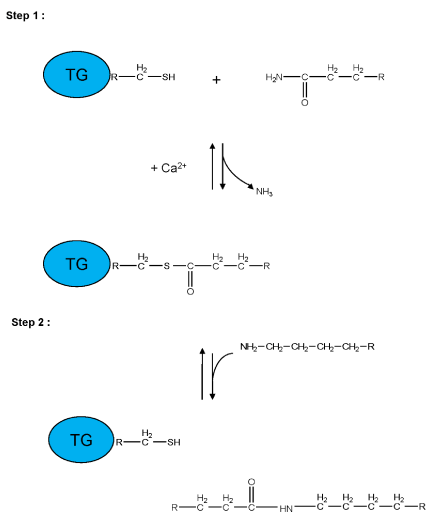
Figure 2: Schematic representation of a two-step transglutaminase reaction. Step 1: In the presence of Ca2+, the active-site cysteine residue reacts with the γ-carboxamide group of an incoming glutaminyl residue of a protein/peptide substrate to yield a thioacyl-enzyme intermediate and ammonia. Step 2: The thioacyl-enzyme intermediate reacts with a nucleophilic primary amine substrate, resulting in the covalent attachment of the amine-containing donor to the substrate glutaminyl acceptor and regeneration of the cysteinyl residue at the active site. If the primary amine is donated by the ε-amino group of a lysyl residue in a protein/polypeptide, a Nε-(γ-L-glutamyl)-L-lysine (GGEL) isopeptide bond is formed.
Ample experimental evidence indicate that some TGs are multifunctional proteins with distinct and regulated enzymatic activities. In fact, under physiological conditions, the transamidation activity of TGs is latent [9,10], while other activities, recently identified, could be present. For example, in some physiological states, when the concentration of Ca2+ increases, the crosslinking activity of TGs may contribute to important biological processes. As previously described, one of the most intriguing properties of some TGs, such as TG2, is the ability to bind and hydrolyze GTP and furthermore, to bind to GTP and Ca2+. GTP and Ca2+ regulate its enzymatic activities, including protein cross-linking, in a reciprocal manner: the binding of Ca2+ inhibits GTP-binding and GTP-binding inhibits the transglutaminase cross-linking activity of the TG2 [5]. Interestingly, TG2 shows no sequence homology with heterotrimeric or low-molecular-weight G-proteins, but there is evidence that TG2 (TG2/Ghα) is involved in signal transduction, and, therefore, TG2/Ghα should also be classified as a large molecular weight G-protein. Other studies, along with ours, showed that TG2/Ghα can mediate the activation of phospholipase C (PLC) by the α1b-adrenergic receptor [10] and can modulate adenylyl cyclase activity [11]. TG2/Ghα can also mediate the activation of the δ1 isoform of PLC and of maxi-K channels [12]. Interestingly, the signaling function of TG2/Ghα is preserved even with the mutagenic inactivation of its crosslinking activity by the mutation of the active site cysteine residue [13].
MOLECULAR BIOLOGY OF THE TRANSGLUTAMINASES
To date at least eight different TGs, distributed in the human body, have been identified (Table 1) [14-19]. Complex gene expression mechanisms regulate the physiological roles that these enzymes play in both the intracellular and extracellular compartments. In the Nervous System, for example, several forms of TGs are simultaneously expressed [20-22]. Moreover, in these last years, several alternative splice variants of TGs, mostly in the 3’-end region, have been identified [23]. Interestingly, some of them are differently expressed in human pathologies, such as Alzheimer’s disease (AD) [24]. On the basis of their ubiquitous expression and their biological roles, we may speculate that the absence of these enzymes would be lethal. However, this does not always seem to be the case, since, for example, null mutants of the TG2 are usually phenotypically normal at birth [12,25,26]. This result may be explained by the expression of other TG genes that may substitute the TG2 missing isoform, although other TG isoform mutations have been associated with severe phenotypes, such as lamellar ichthyosis for TG1 isoform mutations. Bioinformatic studies have shown that the primary structures of human TGs share some identities in only few regions, such as the active site and the calcium binding regions. However, high sequence conservation and, therefore, a high degree of preservation of secondary structure among TG2, TG3 and FXIIIa indicate that these TGs all share four-domain tertiary structures which could be similar to those of other TGs [27].
| Table 1: TGs and their physiological roles when known. | |||
| TG | Physiological role | Gene map location | Reference |
| Factor XIIIa | Blood clotting | 6p24-25 | [14] |
| TG 1 | Skin differentiation | 14q11.2 | [15] |
| (Keratinocyte TG, kTG) TG 2 |
Apoptosis, cell | 20q11-12 | [16] |
| (Tissue TG, tTG, cTG) | adhesion, signal transduction |
||
| TG 3 | Hair follicle | 20p11.2 | [17] |
| (Epidermal TG, eTG) | Differentiation | ||
| TG 4 | Suppression of sperm | 3q21-2 | [18] |
| (Prostate TG, pTG) | Immunogenicity | ||
| TG 5 (TG X) | Epidermal differentiation |
15q15.2 | [19] |
| TG 6 (TG Y) | Central Nervous System Development |
20p13 | [19] |
| TG 7 (TG Z) | Unknown function | 15q15.2 | [19] |
ROLE OF THE TRANSGLUTAMINASES IN NEURODEGENERATIVE DISEASES
Although numerous scientific reports suggest that the transglutaminase activity is involved in the pathogenesis of neurodegenerative diseases, to date, however, still controversial experimental findings about the role of the TGs enzymes in these diseases have been obtained [28-30]. Protein aggregates in affected brain regions are histopathological hallmarks of many neurodegenerative diseases [31]. More than 20 years ago Selkoe et al. [32], suggested that TG activity might contribute to the formation of protein aggregates in AD brain. In support of this hypothesis, tau protein has been shown to be an excellent in vitro substrate of TGs [33,34] and GGEL cross-links have been found in the neurofibrillary tangles and paired helical filaments of AD brains [35]. Interestingly, a recent work showed the presence of bis γ-glutamyl putrescine in human CSF, which was increased in Huntington’s disease (HD) CSF [36]. This is an important evidence that protein/peptides crosslinking by polyamines does indeed occur in the brain, and that this is increased in HD brain. TG activity has been shown to induce also amyloid β-protein oligomerization [37] and aggregation at physiologic levels [38]. By these molecular mechanisms, TGs could contribute to AD symptoms and progression [38]. Moreover, there is evidence that TGs also contribute to the formation of proteinaceous deposits in Parkinson’s disease (PD) [39,40], in supranuclear palsy [41,42] and in HD, a neurodegenerative disease caused by a CAG expansion in the affected gene [43]. For example, expanded polyglutamine domains have been reported to be substrates of TG2 [44-46] and therefore aberrant TG activity could contribute to CAG-expansion diseases, including HD (Figure 3). However, although all these studies suggest the possible involvement of the TGs in the formation of deposits of protein aggregates in neurodegenerative diseases, they do not indicate whether aberrant TG activity per se directly determines the disease progression. For example, several experimental findings reported that TG2 activity in vitro leads to the formation of soluble aggregates of α-synuclein [47] or polyQ proteins [48,49]. To date, as previously reported, at least ten human CAG-expansion diseases have been described (Table 2) [50-59] and in at least eight of them their neuropathology is caused by the expansion in the number of residues in the polyglutamine domain to a value beyond [35-40]. Remarkably, the mutated proteins have no obvious similarities except for the expanded polyglutamine domain. In fact, in all cases except SCA 12, the mutation occurs in the coding region of the gene. However, in SCA12, the CAG triplet expansion occurs in the untranslated region at the 5’ end of the PPP2R2B gene. It has been proposed that the toxicity results from overexpression of the brain specific regulatory subunit of protein phosphatase PP2A [56]. Most of the mutated proteins are widely expressed both within the brain and elsewhere in the body. A major challenge then is to understand why the brain is primarily affected and why different regions within the brain are affected in the different CAG-expansion diseases, i.e., what accounts for the neurotoxic gain of function of each protein and for a selective vulnerability of each cell type. Possibly, the selective vulnerability [60] may be explained in part by the susceptibility of the expanded polyglutamine domains in the various CAG-expansion diseases to act as cosubstrates for a brain TG (Figure 4). To strengthen the possible central role of the TGs in neurodegenerative diseases, a study by Hadjivassiliou et al. [61], showed that anti-TG2 IgA antibodies are present in the gut and brain of patients with gluten ataxia, a non-genetic sporadic cerebellar ataxia, but not in ataxia control patients. Recently, anti-TG2, -TG3 and -TG6 antibodies have been found in sera from CD patients, suggesting a possible involvement also of other TGs in the pathogenesis of dermatitis herpetiformis and gluten ataxia, two frequent extra intestinal manifestations of gluten sensitivity [62,63]. These last findings could suggest also a possible role of the “gut-brain axe” for the etiopathogenesis of human neurodegenerative diseases, in which the TG enzymes, in particular the TG2 enzyme, could play an important role [64-66].
| Table 2: List of polyglutamine (CAG-expansion) diseases. | |||||
| Disease | Sites of neuropathology | CAG triplet number | Gene product (Intracellular localization of protein deposits) | Reference | |
| Normal | Disease | ||||
| Corea Major or Huntington’s Disease (HD) | Striatum (medium spiny neurons) and cortex in late stage | 6–35 | 36–121 | Huntingtin(n,c) | [50] |
| Spinocerebellar Ataxia Type 1 (SCA1) | Cerebellar cortex (Purkinje cells), dentate nucleus and brain stem | 6–39 | 40–81 | Ataxin-1(n,c) | [51] |
| Spinocerebellar Ataxia Type 2 (SCA2) | Cerebellum, pontine nuclei, substantia nigra | 15–29 | 35–64 | Ataxin–2 (c) | [52] |
| Spinocerebellar Ataxia Type 3 (SCA3) or Machado-Joseph disease (MJD) | Substantia nigra, globus pallidus, pontine nucleus, cerebellar cortex | 13–42 | 61–84 | Ataxin –3 (c) | [53] |
| Spinocerebellar Ataxia Type 6 (SCA6) | Cerebellar and mild brainstem atrophy | 4–18 | 21–30 | Calcium channel Subunit (a 1A)(m) | [54] |
| Spinocerebellar Ataxia Type 7 (SCA7) | Photoreceptor and bipolar cells, cerebellar cortex, brainstem | 7–17 | 37–130 | Ataxin-7 (n) | [55] |
| Spinocerebellar Ataxia Type 12 (SCA12) | Cortical, cerebellar atrophy | 7–32 | 41–78 | Brain specific regulatory subunit of protein phosphatase PP2A (?) | [56] |
| Spinocerebellar Ataxia Type 17 (SCA17) | Gliosis and neuronal loss in the Purkinje cell layer | 29–42 | 46–63 | TATA-binding protein (TBP) (n) | [57] |
| Spinobulbar Muscular Atrophy (SBMA) or Kennedy Disease | Motor neurons (anterior horn cells, bulbar neurons) and dorsal root ganglia | 11–34 | 40–62 | Androgen receptor (n, c) | [58] |
| Dentatorubral-pallidoluysian Atrophy (DRPLA) | Globus pallidus, dentato-rubral and subthalamic nucleus | 7–35 | 49–88 | Atrophin (n, c) | [59] |
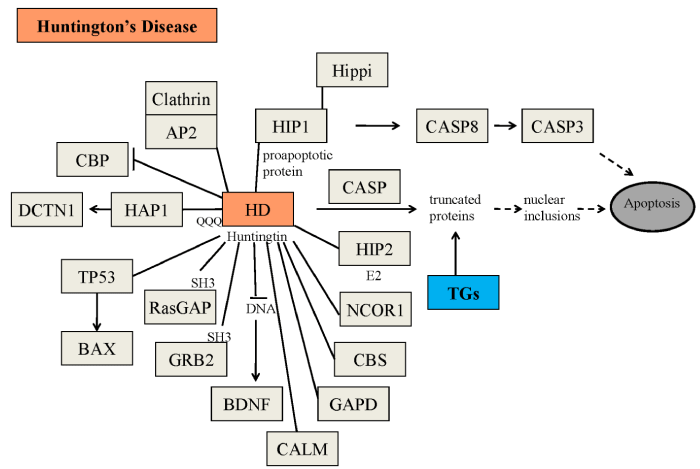
Figure 3: Possible physiopathological effects of the mutated huntingtin. Some of the physiopathological roles of mutated huntingtin, including the formation of nuclear inclusions, have been described in the Figure AP2=adipocyte Protein 2; BAX=bcl-2-like protein 4; BDNF=brain-derived neurotrophic factor; CALM= calmodulin; CASP=caspases; CASP3=caspase 3; CASP8=caspase 8; CBP= CREB binding protein; CBS = cystathionine-β-synthase; DCTN1 = dynactin subunit 1; GAPD=glyceraldehyde-3-phosphate dehydrogenase; GRB2=growth factor receptor-bound protein 2; HAP1=huntingtin associated protein 1; HIP1=huntingtin interacting protein 1; HIP2=huntingtin interacting protein 2; Hippi; HIP1 protein interactor; NCOR1=nuclear receptor corepressor 1; RasGAP=p21Ras protein and GTPase-activating protein complex; TGs=transglutaminases; TP53=tumor protein 53.
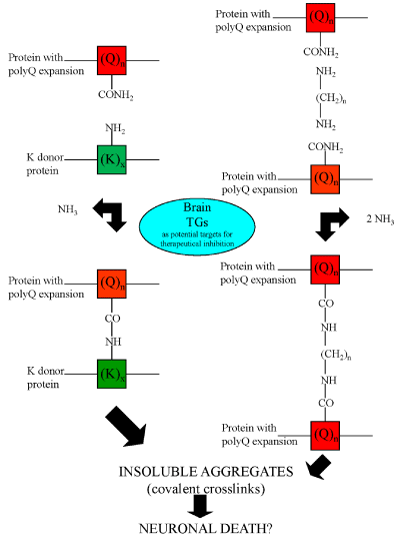
Figure 4: Possible mechanisms responsible for protein aggregate formation catalyzed by TGs. Transglutaminase activity could produce insoluble aggregates both by the formation of Nε-(γ-L-glutamyl)-L-lysine (GGEL) isopeptide bonds (left side of the figure) and by the formation of N,N-bis-(γ-L-glutamyl)polyamine bridges (right side of the figure) in the mutated huntingtin.
In support of the hypothesis of the toxic effect of TG activity in other neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, TG activity has been shown to induce amyloid beta-protein and α-synuclein oligomerization and aggregation at physiologic levels [67-69]. In fact, TG activity induces protofibril-like amyloid beta-protein assemblies that are protease-resistant and inhibit long-term potentiation [38]. Therefore, by these molecular mechanisms, TG activity could also contribute to Alzheimer’s disease symptoms and progression. Very recently, TG2 and its product isopeptide have been found increased in Alzheimer’s disease and APPswe/PS1dE9 double transgenic mice brains [70], while catalytically active TG2 colocalizes with Aβ pathology in Alzheimer’s disease mouse models [71]. Interestingly, other works are suggesting that also other TGs could be involved in the molecular mechanisms responsible for neurodegenerative diseases [72]. In particular, a recent work by Basso et al. [73], found that in addition to TG2, TG1 gene expression level is significantly induced following stroke in vivo or due to oxidative stress in vitro. Moreover, structurally diverse inhibitors, used at concentrations that inhibit TG1 and TG2 simultaneously, are neuroprotective. Together, these last studies suggested that multiple TG isoforms, not only TG2, participate in oxidative stress-induced cell death signalling, and that isoform nonselective inhibitors of TG will be most efficacious in combating oxidative death in neurological disorders. These are interesting and worthwhile studies, suggesting that multiple TG isoforms can participate in neuronal death processes. Therefore, all these studies suggest that the involvement of brain TGs could represent a common denominator in several neurological diseases, which can lead to the determination of pathophysiological consequences through different molecular mechanisms.
ROLE OF THE TRANSGLUTAMINASE ACTIVITY IN NEUROINFLAMMATION
Neuroinflammation plays an important role in various chronic neurodegenerative diseases, characterized also by the pathological accumulation of specific protein aggregates. In particular, several of these proteins have been shown to be substrates of transglutaminases. Interestingly, it has recently been demonstrated that transglutaminase 2 (TG2) may also be involved in molecular mechanisms underlying inflammation. In the central nervous system, astrocytes and microglia are the cell types mainly involved in this inflammatory process. The transcription factor NF-κB is considered the main regulator of inflammation and it is activated by a variety of stimuli including calcium influx, oxidative stress and inflammatory cytokines. Recently, in addition to these stimuli, TG2 has been shown to activate NF-κB both via a canonical pathway [74] and via a non-canonical pathway [75]. On the other hand, NF-κB regulatory response elements are present also in the Transglutaminase 2 promoter [76]. Under these conditions, the over-expression of TG2 results in the sustained activation of NF-κB. Several findings emphasize the possible role of the TG2/NF-κB activation pathway in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, multiple sclerosis and amyotrophic lateral sclerosis. Together, these evidences suggest that TG2 could play a role in neuroinflammation and could contribute to the production of compounds that are potentially deleterious to neuronal cells [77].
TRANSGLUTAMINASE INHIBITION AS POSSIBLE THERAPEUTICAL APPROACH
In consideration to the fact that up to now there have been no long-term effective treatments for the human neurodegenerative diseases previously reported, then the possibility that selective TG inhibitors may be of clinical benefit has been seriously considered. In this respect, some encouraging results have been obtained with TG inhibitors in preliminary studies with different biological models of CAG-expansion diseases. For example, cystamine (Figure 5) is a potent in vitro inhibitor of enzymes that require an unmodified cysteine at the active site [78]. Inasmuch as TGs contain a crucial active-site cysteine, cystamine has the potential to inhibit these enzymes by a sulfide-disulfide interchange reaction. A sulfide-disulfide interchange reaction results in the formation of cysteamine and a cysteamine-cysteine mixed disulfide residue at the active site. Recent studies have shown that cystamine decreases the number of protein inclusions in transfected cells expressing the atrophin (DRPLA) protein containing a pathological-length polyglutamine domain [79]. In other studies, cystamine administration to HD-transgenic mice resulted in an increase in life expectancy and amelioration of neurological symptoms [80,81]. Neuronal inclusions were decreased in one of these studies [81]. Although all these scientific reports seem to support the hypothesis of a direct role of transglutaminase activity in the pathogenesis of the polyglutamine diseases, cystamine is also found to act in the HD-transgenic mice by mechanisms other than the inhibition of TGs, such as the inhibition of caspases [82], suggesting that this compound can have an additive effect in the therapy of HD. Currently, cysteamine is already in phase I studies in humans with HD [83], but several side effects, such as nausea, motor impairment and dosing schedule have been reported as reasons for non-adherence during phase II studies in human patients affected by cystinosis [84,85]. Another critical problem in the use of TG inhibitors in treating neurological diseases relates to the fact that, as previously reported, the human brain contains at least four TGs, including TG1,2,3 [22] and TG6 [86], and a strong non-selective inhibitor of TGs might also inhibit plasma Factor XIIIa, causing a bleeding disorder. Therefore, from a number of standpoints it would seem that a selective inhibitor, which discriminates between TGs, would be preferable to an indiscriminate TG inhibitor. In fact, although most of the TG activity in mouse brain, at least as assessed by an assay that measures the incorporation of radioactive putrescine (amine donor) into N,N-dimethyl casein (amine acceptor), seems to be due to TG2 [87], no conclusive data have been obtained by TG2 gene knock-out experiments about the involvement of this TG in the development of the symptoms in HD-transgenic mice [26,88,89]. Moreover, a recent scientific report showed that cystamine reduces aggregate formation in a mouse model of oculopharyngeal muscular dystrophy (OMPD), in which also the TG2 knockdown is capable of suppressing the aggregation and the toxicity of the mutant protein PABPN1 [90], suggesting this compound as a possible therapeutic for OMPD.
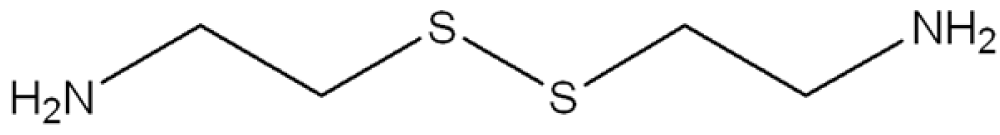
Figure 5: Chemical structure of cystamine.
CONCLUSION
Numerous scientific reports have implicated aberrant TG activity in neurodegenerative diseases, but still today we are looking for experimental findings which could definitely confirm the direct involvement of TGs in the pathogenetic mechanisms responsible for these diseases. However, as result of the putative role of specific TG isoforms, such as TG2, in some human diseases, there is a considerable interest in developing inhibitors of these enzymes. Of those currently available, cystamine is the most commonly used experimentally to inhibit TG2 activity. In addition to cystamine, several types of TG2 inhibitors have been developed up to now [91]. Interestingly, some of these inhibitors have shown promising results in experimental diabetic models [92]. Therefore, the use of these inhibitors of TGs could be then useful also for other clinical approaches. To minimize the possible side effects, however, more selective inhibitors of the TGs should be required in the future. Progress in this area of research could be achieved, if possible, also through pharmaco-genetic approaches.
ACKNOWLEDGEMENTS
This work is supported by the Italian Education Department and the Regione Campania (L.R. n.5 del 28.03.2002, finanziamento 2008) entitled: Identificazione e caratterizzazione di geni della transglutaminasi nel Sistema Nervoso in relazione allo sviluppo di malattie neurodegenerative (Identification and characterization of transglutaminase genes in the Nervous System in relationship to the development of neurodegenerative diseases).
REFERENCES
- Folk JE. Mechanism and basis for specificity of transglutaminase-catalyzed e-(g-glutamyl) lysine bond formation. Adv Enzymol Relat Areas Mol Biol. 1983; 54: 1-56. Ref.: https://goo.gl/MZtXQS
- Lorand L, Conrad S M. Transglutaminases. Mol Cell Biochem. 1984; 58: 9-35. Ref.: https://goo.gl/uAd6qy
- Piacentini M, Martinet N, Beninati S, Folk JE. Free and protein conjugated-polyamines in mouse epidermal cells. Effect of high calcium and retinoic acid. J Biol Chem. 1988; 263: 3790-3794. Ref.: https://goo.gl/47QA8F
- Song Y, Kirkpatrick LL, Schilling AB, Helseth DL, Chabot N, et al. Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron. 2013; 78: 109-123. Ref.: https://goo.gl/8xamTn
- Achyuthan KE, Greenberg CS. Identification of a guanosine triphosphate-binding site on guinea pig liver transglutaminase. Role of GTP and calcium ions in modulating activity. J Biol Chem. 1987; 262: 1901-1906. Ref.: https://goo.gl/GVXy9h
- Hasegawa G, Suwa M, Ichikawa Y, Ohtsuka T, Kumagai S, et al. A novel function of tissue-type transglutaminase: protein disulfide isomerase. Biochem J. 2003; 373: 793-803. Ref.: https://goo.gl/J7PR9u
- Lahav J, Karniel E, Bagoly Z, Sheptovitsky V, Dardik R, et al. Coagulation factor XIII serves as protein disulfide isomerase. Thromb Haemost. 2009; 101: 840-844. Ref.: https://goo.gl/sCnzMA
- Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev. 2009; 89: 991-1023. Ref.: https://goo.gl/H9sRQC
- Smethurst PA, Griffin M. Measurement of tissue transglutaminase activity in a permeabilized cell system: its regulation by calcium and nucleotides. Biochem J. 1996; 313: 803-808. Ref.: https://goo.gl/cVYj3d
- Nakaoka H, Perez DM, Baek KJ, Das T, Husain A, et al. Gh: a GTP-binding protein with transglutaminase activity and receptor signalling function. Science 1994; 264: 1593-1596. Ref.: https://goo.gl/NdY8xz
- Gentile V, Porta R, Chiosi E, Spina A, Valente F, et al. tTGase/G alpha h protein expression inhibits adenylate cyclase activity in Balb-C 3T3 fibroblasts membranes. Biochim Biophys Acta. 1997; 1357: 115-122. Ref.: https://goo.gl/zz6rVF
- Nanda N, Iismaa SE, Owens WA, et al. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001; 276: 20673-20678. Ref.: https://goo.gl/nvfyq4
- Mian S, El Alaoui S, Lawry J, Gentile V, Davies PJ, et al. The importance of the GTP binding protein tissue transglutaminase in the regulation of cell cycle progression. FEBS Letters. 1995; 370: 27-31. Ref.: https://goo.gl/ddWehz
- Olaisen B, Gedde-Dahl TJR, Teisberg P, Thorsby E, Siverts A, et al. A structural locus for coagulation factor XIIIA (F13A) is located distal to the HLA region on chromosome 6p in man. Am J Hum Genet. 1985; 37: 215-220. Ref.: https://goo.gl/NvA1Kn
- Yamanishi K, Inazawa J, Liew FM, Nonomura K, Ariyama T, et al. Structure of the gene for human transglutaminase 1. J Biol Chem. 1992; 267: 17858-17863. Ref.: https://goo.gl/JMjQWB
- Gentile V, Davies PJ, Baldini A. The human tissue transglutaminase gene maps on chromosome 20q12 by in situ fluorescence hybridization. Genomics. 1994; 20: 295-297. Ref.: https://goo.gl/oW3hdr
- Wang M, Kim IG, Steinert PM, McBride OW. Assignment of the human transglutaminase 2 (TGM2) and transglutaminase 3 (TGM3) genes to chromosome 20q11.2. Genomics. 1994; 23: 721-722. Ref.: https://goo.gl/AArGWB
- Gentile V, Grant F, Porta R.. Human prostate transglutaminase is localized on chromosome 3p21.33-p22 by in situ fluorescence hybridization. Genomics. 1995; 27: 219-220.
- Grenard P, Bates MK, Aeschlimann D. Evolution of transglutaminase genes: identification of a transglutaminases gene cluster on human chromosome 15q. Structure of the gene encoding transglutaminase X and a novel gene family member, transglutaminase Z. J Biol Chem. 2001; 276: 33066-33078. Ref.: https://goo.gl/Hpkd7L
- Thomas H, Beck K, Adamczyk M, Aeschlimann P, Langley M, et al. Transglutaminase 6: a protein associated with central nervous system development and motor function. Amino Acids. 2013; 44: 161-177. Ref.: https://goo.gl/ghKXLi
- Bailey CD, Johnson GV. Developmental regulation of tissue transglutaminase in the mouse forebrain. J Neurochem. 2004; 91: 1369-1379. Ref.: https://goo.gl/vyNVAK
- Kim SY, Grant P, Lee JH, Pant HC, Steinert PM. Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminase 1 and 2 in Alzheimer’s disease. J Biol Chem. 1999; 274: 30715-30721. Ref.: https://goo.gl/WYtcCc
- lannaccone M, Giuberti G, De Vivo G, Caraglia M, Gentile V. Identification of a FXIIIA variant in human neuroblastoma cell lines. Int J Biochem Mol Biol. 2013; 4: 102-107. Ref.: https://goo.gl/jiPkov
- Citron BA, Santa Cruz KS, Davies PJ, Festoff BW. Intron-exon swapping of transglutaminase mRNA and neuronal tau aggregation in Alzheimer’s disease. J Biol Chem. 2001; 276: 3295-3301. Ref.: https://goo.gl/kaoiR9
- De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol 2001; 21: 148–155. Ref.: https://goo.gl/EaYkTA
- Mastroberardino PG, Iannicola C, Nardacci R, Bernassola F, De Laurenzi V, et al. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 2002; 9: 873-880. Ref.: https://goo.gl/UFGgnZ
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nature Mol Cell Biol. 2003; 4: 140-156. Ref.: https://goo.gl/uB25U4
- Wolf J, Jäger C, Lachmann I, Schönknecht P, Morawski M, et al. Tissue transglutaminase is not a biochemical marker for Alzheimer’s disease. Neurobiol Aging. 2013; 34: 2495-2498. Ref.: https://goo.gl/k7fpBu
- Wilhelmus MM, Drukarch B. Tissue transglutaminase is a biochemical marker for Alzheimer’s disease. Neurobiol Aging. 2014; 35: 3-4. Ref.: https://goo.gl/5HCQ3p
- Wolf J, Jäger C, Morawski M, Lachmann I, Schönknecht P, et al. Tissue transglutaminase in Alzheimer’s disease-facts and fiction: a reply to “Tissue transglutaminase is a biochemical marker for Alzheimer’s disease”. Neurobiol Aging. 2014; 35: 5-9. Ref.: https://goo.gl/n6iGhe
- Adams RD, Victor M. Principles of Neurology. McGraw-Hill, Inc. Ed. 1993.
- Selkoe DJ, Salazar FJ, Ihara Y. Alzheimer’s disease: insolubility of partially purified paired helical filaments in sodium dodecyl sulfate and urea. Proc Natl Acad Sci USA. 1982; 215: 1243-1245. Ref.: https://goo.gl/6ttFbK
- Grierson AJ, Johnson GV, Miller CC. Three different human isoforms and rat neurofilament light, middle and heavy chain proteins are cellular substrates for transglutaminase. Neurosci Lett. 2001; 298: 9-12. Ref.: https://goo.gl/n5qJcb
- Singer SM, Zainelli GM, Norlund MA, Lee JM, Muma NA. Transglutaminase bonds in neurofibrillary tangles and paired helical filament t early in Alzheimer’s disease. Neurochem Int. 2002; 40: 17-30. Ref.: https://goo.gl/6sJKUe
- Halverson RA, Lewis J, Frausto S, Hutton M, Muma NA. Tau protein is cross-linked by transglutaminase in P301L tau transgenic mice. J Neurosci. 2005; 25: 1226-1233. Ref.: https://goo.gl/XgxnaL
- Jeitner TM, Matson WR, Folk JE, Blass JP, Cooper AJ. Increased levels of g-glutamylamines in Huntington disease CSF. J Neurochem. 2008; 106: 37-44. Ref.: https://goo.gl/uk1ek3
- Dudek SM, Johnson GV. Transglutaminase facilitates the formation of polymers of the beta-amyloid peptide. Brain Res 1994; 651: 129-133. Ref.: https://goo.gl/fJ6Y6h
- Hartley DM, Zhao C, Speier AC, Woodard GA, Li S, et al. Transglutaminase induces protofibril-like amyloid b protein assemblies that are protease-resistant and inhibit long-term potentiation. J Biol Chem. 2008; 283: 16790-16800. Ref.: https://goo.gl/3Ntz4n
- Citron BA, Suo Z, SantaCruz K, Davies PJ, Qin F, et al. Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int. 2002; 40: 69-78. Ref.: https://goo.gl/5PLq8j
- Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM. Tissue transglutaminase-induced aggregation of a-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 2003; 100: 2047-2052. Ref.: https://goo.gl/Ch8MFv
- Zemaitaitis MO, Lee JM, Troncoso JC, Muma NA. Transglutaminase-induced cross-linking of t proteins in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2000; 59: 983-989. Ref. : https://goo.gl/Uy1D3R
- Zemaitaitis MO, Kim SY, Halverson RA, Troncoso JC, Lee JM, et al. Transglutaminase activity, protein, and mRNA expression are increased in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2003 ; 62: 173-184. Ref. : https://goo.gl/2fVehR
- Iuchi S, Hoffner G, Verbeke P, Djian P, Green H. Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc Natl Acad Sci USA. 2003; 100: 2409-2414. Ref.: https://goo.gl/iAgP4t
- Gentile V, Sepe C, Calvani M, Melone MA, Cotrufo R, et al. Tissue transglutaminase-catalyzed formation of high-molecular-weight aggregates in vitro is favored with long polyglutamine domains: a possible mechanism contributing to CAG-triplet diseases. Arch Biochem Biophys. 1998; 352: 314-321. Ref.: https://goo.gl/PV65Tc
- Kahlem P, Green H, Djian P. Transglutaminase action imitates Huntington’s disease: selective polymerization of huntingtin containing expanded polyglutamine. Mol Cell 1998; 1: 595-601. Ref.: https://goo.gl/WmmuSY
- Karpuj MV, Garren H, Slunt H, Price DL, Gusella J, et al. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington’s disease brain nuclei. Proc Natl Acad Sci USA. 1999; 96: 7388-7393. Ref.: https://goo.gl/VYQi2k
- Segers-Nolten IM, Wilhelmus MM, Veldhuis G, Benjamin Drukarch, Benjamin Drukarch, subramaniam V, et al. Tissue transglutaminase modulates a-synuclein oligomerization. Protein Sci. 2008 ; 17: 1395–1402. Ref. : https://goo.gl/unCQRM
- Lai TS, Tucker T, Burke JR, Strittmatter WJ, Greenberg CS. Effect of tissue transglutaminase on the solubility of proteins containing expanded polyglutamine repeats. J Neurochem. 2004; 88: 1253-1260. Ref.: https://goo.gl/S1J74t
- Konno T, Mori T, Shimizu H, et al.Paradoxical inhibition of protein aggregation and precipitation by transglutaminase-catalyzed intermolecular cross-linking. J Biol Chem. 2005; 280: 17520-17525. Ref.: https://goo.gl/Ry5GEK
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosome. Cell. 1993; 72: 971-983. Ref.: https://goo.gl/qmv1AP
- Banfi S, Chung MY, McCall AE, Chinault AC, Zoghbi HY, et al. Mapping and cloning of the critical region for the spinocerebellar ataxia type 1 gene (SCA1) in a yeast artificial chromosome contig spanning 1.2 Mb. Genomics. 1993; 18: 627-635. Ref.: https://goo.gl/ZvWKTJ
- Sanpei K, Takano H, Igarashi S, Oyake M, Ishida Y, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996; 14: 277-284. Ref.: https://goo.gl/FzpaFG
- Pujana MA, Volpini V, Estivill X. Large CAG/CTG repeat templates produced by PCR, usefulness for the DIRECT method of cloning genes with CAG/CTG repeat expansions. Nucleic Acids Res. 1998; 1: 1352-1353. Ref.: https://goo.gl/5GLDuz
- Fletcher CF, Lutz CM, O’Sullivan TN, Shaughnessy JD Jr, Hawkes R, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996; 87: 607-617. Ref.: https://goo.gl/7q7r2Y
- Vincent JB, Neves-Pereira ML, Paterson AD, Yamamoto E, Parikh SV, et al. An unstable trinucleotide-repeat region on chromosome 13 implicated in spinocerebellar ataxia: a common expansion locus. Am J Hum Genet. 2000; 66: 819-829. Ref.: https://goo.gl/nBXuv9
- Holmes SE, O’Hearn E, Margolis RL. Why is SCA12 different from other SCAs? Cytogenet Genome Res. 2003; 100: 189-197. Ref.: https://goo.gl/vZNHmg
- Imbert G, Trottier Y, Beckmann J, Mandel JL.The gene for the TATA binding protein (TBP) that contains a highly polymorphic protein coding CAG repeat maps to 6q27. Genomics. 1994; 21: 667-668. Ref.: https://goo.gl/T7vNi1
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991; 352: 77-79. Ref.: https://goo.gl/VLB1KC
- Onodera O, Oyake M, Takano H, Ikeuchi T, Igarashi S, et al. Molecular cloning of a full-length cDNA for dentatorubral-pallidoluysian atrophy and regional expressions of the expanded alleles in the CNS. Am J Hum Genet. 1995; 57: 1050-1060. Ref.: https://goo.gl/AeuaVm
- Cooper AJL, Sheu K-FR, Burke JR, Strittmatter WJ, Gentile V, et al. Pathogenesis of inclusion bodies in (CAG) n/Qn-expansion diseases with special reference to the role of tissue transglutaminase and to selective vulnerability. J Neurochem. 1999; 72: 889-899. Ref.: https://goo.gl/EqLyLf
- Hadjivassiliou M, Maki M, Sanders DS, Williamson CA, Grünewald RA, et al.Autoantibody targeting of brain and intestinal transglutaminase in gluten ataxia. Neurology. 2006; 66: 373-377. Ref.: https://goo.gl/26qTHDc
- Boscolo S, Lorenzon A, Sblattero D, Florian F, Stebel M, et al. Anti-transglutaminase antibodies cause ataxia in mice. Plos One. 2010; 5: 9698. Ref.: https://goo.gl/gnQJgY
- Stamnaes J, Dorum S, Fleckenstein B, Aeschlimann D, Sollid LM. Gluten T cell epitope targeting by TG3 and TG6; implications for dermatitis herpetiformis and gluten ataxia. Amino Acids. 2010; 39: 1183-1191. Ref.: https://goo.gl/nMey3w
- Lerner A, Matthias T. GUT-the Trojan horse in remote organs’ autoimmunity. J Clin Cell Immunol. 2016; 7: 401.
- Matthias T, Jeremias P, Neidhofer S, Lerner A. The industrial food additive microbial transglutaminase, mimics the tissue transglutaminase and is immunogenic in celiac disease patients. Autoimmun Rev. 2016; 15: 1111-1119. Ref.: https://goo.gl/6BtgYn
- Lerner A, Neidhofer S, Matthias T. Transglutaminase 2 and anti-transglutaminase 2 autoantibodies in celiac disease and beyond: Part A: TG2 double-edged sword: gut and extraintestinal involvement. Immunome Res. 2015; 11: 101-105.
- Wakshlag JJ, Antonyak MA, Boehm JE, Boehm K, Cerione RA. Effects of tissue transglutaminase on beta-amyloid 1-42-induced apoptosis. Protein J 2006; 25: 83-94. Ref.: https://goo.gl/sbpHZh
- Lee JH, Jeong J, Jeong EM, Cho SY, Kang JW, et al. Endoplasmic reticulum stress activates transglutaminase 2 leading to protein aggregation. Int J Mol Med. 2014; 33: 849-855. Ref.: https://goo.gl/RKBjLo
- Grosso H, Woo JM, Lee KW, Im JY, Masliah E, et al. Transglutaminase 2 exacerbates α-synuclein toxicity in mice and yeast. FASEB J. 2014; 28: 4280-4291. Ref.: https://goo.gl/bQWkee
- Zhang J, Wang S, Huang W, Bennett DA, Dickson DW, et al. Tissue transglutaminase and its product isopeptide are increased in Alzheimer’s disease and APPswe/PS1dE9 double transgenic mice brains. Mol Neurobiol. 2016; 53: 5066-5078. Ref.: https://goo.gl/FrtFxH
- Wilhelmus MM, De JM, Smit AB, van der Loo RJ, Drukarch B. Catalytically active tissue transglutaminase colocalises with Ab pathology in Alzheimer’s disease mouse models. Sci Rep. 2016; 6: 20569. Ref.: https://goo.gl/9CXX9G
- Wilhelmus MMM, De JM, Rozemuller AJM, Brevé J, Bol JG, et al.Transglutaminase 1 and its regulator Tazarotene-induced gene 3 localize to neuronal tau inclusions in tauopathies. J Pathol. 2012; 226: 132-142. Ref.: https://goo.gl/1bNwTs
- Basso M, Berlin J, Xia L, Sleiman SF, Ko B, et al. Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci. 2012; 39: 6561-6569. Ref.: https://goo.gl/RgYNz5
- Lee J, Kim YS, Choi DH, Bang MS, Han TR, et al.Transglutaminase 2 induces nuclear factor-kB activation via a novel pathway in BV-2 microglia. J Biol Chem. 2004; 279: 53725-53735. Ref.: https://goo.gl/4YD22W
- Kumar S, Mehta K. Tissue transglutaminase constitutively activates HIF-1a promoter and nuclear factor-kB via a non-canonical pathway. Plos One. 2012; 7: 49321. Ref.: https://goo.gl/s4TNqZ
- Lu S, Saydak M, Gentile V, Stein JP, Davies PJ. Isolation and characterization of the human tissue transglutaminase promoter. J Biol Chem. 1995; 270: 9748-9755. Ref.: https://goo.gl/Zd4Psg
- Ientile R, Currò M, Caccamo D. Transglutaminase 2 and neuroinflammation. Amino Acids. 2015; 47: 19-26. Ref.: https://goo.gl/pVvQY8
- Griffith OW, Larsson A, Meister A. Inhibition of g-glutamylcysteine synthetase by cystamine: an approach to a therapy of 5-oxoprolinuria (pyroglutamic aciduria). Biochem Biophys Res Commun. 1977; 79: 919-925. Ref.: https://goo.gl/Jh9aYN
- Igarashi S, Koide R, Shimohata T, Yamada M, Hayashi Y, et al. Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA protein with an expanded polyglutamine stretch. Nat Genet. 1998; 18: 111-117. Ref.: https://goo.gl/jUNMtb
- Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, et al.Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002; 8: 143-149. Ref.: https://goo.gl/rndteF
- Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, et al. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J Neurosci. 2002; 22: 8942-8950. Ref.: https://goo.gl/HFN37j
- Lesort M, Lee M, Tucholski J, Johnson GV. Cystamine inhibits caspase activity. Implications for the treatment of polyglutamine disorders. J Biol Chem. 2003; 278: 3825-3830. Ref.: https://goo.gl/WvVYVR
- Dubinsky R, Gray C. CYTE-I-HD: Phase I dose finding and tolerability study of Cysteamine (Cystagon) in Huntington’s disease. Movement Disord. 2006; 21: 530-533. Ref.: https://goo.gl/8tVNpk
- Langman CB, Greenbaum LA, Sarwal M, Grimm P, Niaudet P, et al. A randomized controlled crossover trial with delayed-release cysteamine bitartrate in nephropathic cystinosis: effectiveness on white blood cell cystine levels and comparison of safety. Clin J Am Soc Nephrol. 2012; 7: 1112-1120. Ref.: https://goo.gl/cdgC1S
- Besouw M, Masereeuw R, Van DHL, Levtchenko E. Cysteamine: an old drug with new potential. Drug Discov Today. 2013; 18: 785-792. Ref.: https://goo.gl/57XnyQ
- Hadjivassiliou M, Aeschlimann P, Strigun A, Sanders DS, Woodroofe N, et al. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann Neurol. 2008; 64: 332-343. Ref.: https://goo.gl/6nc1rD
- Krasnikov BF, Kim SY, McConoughey SJ, Ryu H, Xu H, et al. Transglutaminase activity is present in highly purified nonsynaptosomal mouse brain and liver mitochondria. Biochemistry. 2005; 44: 7830-7843. Ref.: https://goo.gl/ZoXaCr
- Liliana B. Menalled, Kudwa AE, Oakeshott S, Andrew Farrar, Neil Paterson, et al. Genetic deletion of transglutaminase 2 does not rescue the phenotypic deficits observed in R6/2 and zQ175 mouse models of Huntington’s disease. Plos One. 2014; 9: 99520. Ref.: https://goo.gl/pF1NcS
- Bailey CD, Johnson GV. Tissue transglutaminase contributes to disease progression in the R6/2 Huntington’s disease mouse model via aggregate-independent mechanisms. J Neurochem. 2005; 92: 83-92. Ref.: https://goo.gl/iX3VfA
- Davies JE, Rose C, Sarkar S, Rubinsztein DC.Cystamine suppresses polyalanine toxicity in a mouse model of oculopharyngeal muscular dystrophy. Sci Transl Med. 2010; 2: 34-40. Ref.: https://goo.gl/HcNjQ3
- Pietsch M, Wodtke R, Pietzsch J, Löser R. Tissue transglutaminase: An emerging target for therapy and imaging. Bioorg Med Chem Lett. 2013; 23: 6528-6543. Ref.: https://goo.gl/Shgc3j
- Bhatt MP, Lim YC, Hwang J, Na S, Kim YM, et al. C-peptide prevents hyperglycemia-induced endothelial apoptosis through inhibition of reactive oxygen species-mediated transglutaminase 2 activation. Diabetes. 2013; 62: 243-253. Ref.: https://goo.gl/U1EbFJ